Sections
Zymo Mock Community Analysis
ATCC Mock Human Communities Analysis
Madison Water Samples Analysis
Computing Environment
R v3.6.1
- Bioconductor v3.10
- DADA2 v1.14.0
- ShortRead v1.44.0
- ggplot2 v3.2.1
CentOS6 Linux
- Bowtie v2.2.9
- samtools v1.3.1
- Alfred v0.1.17
- Kraken v2.0.8-beta
- Bracken v2.5
- trimmomatic v0.36
- FLASH v1.2.11
- cutadapt v1.16
Zymo Mock Community Analysis
The Zymo mock community was analyzed using three methods:
- Mapping LoopSeq 16S contigs and Illumina V3V4 short reads to provided references
- Mapping LoopSeq 16S contigs to NCBI RefSeq references
- Classifying LoopSeq 16S contigs with LCA classifier
Data Preparation
Mock Community Description
The ZymoBIOMICS Microbial Community DNA Standard is composed of ten microbial species: eight bacteria and two fungi, the latter two of which were not amplified by the LoopSeq 16S rRNA primers used. Genomic DNA quantity from the eight species are evenly distributed and vary in Gram stain, genome size, GC content, 16S copy number and phylogenetic relatedness.
| Species | 16S copies | Expected contig % |
|---|---|---|
| Bacillus subtilis | 10 | 17.4 |
| Enterococcus faecalis | 4 | 9.9 |
| Escherichia coli | 7 | 10.1 |
| Lactobacillus fermentum | 5 | 18.4 |
| Listeria monocytogenes | 6 | 14.1 |
| Pseudomonas aeruginosa | 4 | 4.2 |
| Salmonella enterica | 7 | 10.4 |
| Staphylococcus aureus | 6 | 15.5 |
LoopSeq 16S Contig QC & Filtering
Load fastq file containing raw contigs as downloaded from the Loop Genomics cloud platform.
#R
library(dada2)
library(ShortRead)
F_prim <- "AGAGTTTGATCMTGGCTCAG" #forward primer
R_prim <- "TACCTTGTTACGACTT" #reverse primer
zymo <- file.path("zymo.fastq")
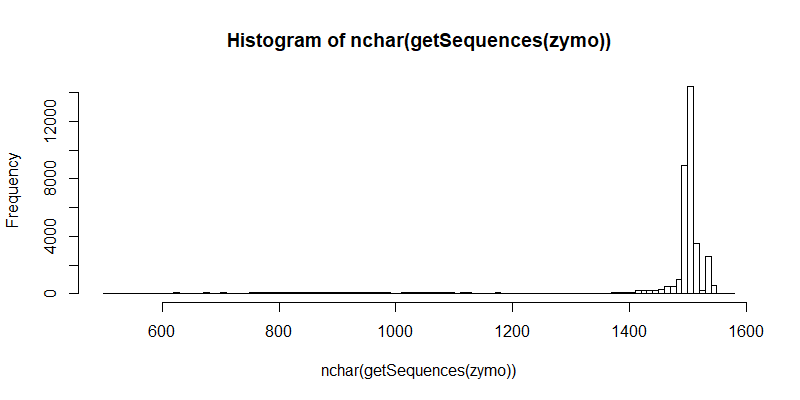
#Basic statistics of raw contigs
summary(nchar(getSequences(zymo)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
500 1494 1508 1430 1510 1572
#Distribution of raw contigs
hist(nchar(getSequences(zymo)), 100)
#Remove forward and reverse primers. This step also ensures that the contigs that remain after filtering are the full length 16S gene.
removePrimers(zymo, paste0("zymo_noprimers.fastq"), primer.fwd=F_prim, primer.rev=rc(R_prim), compress=FALSE, max.mismatch=2, allow.indels=FALSE, verbose=TRUE)
Read in 38629, output 27123 (70.2%) filtered sequences.
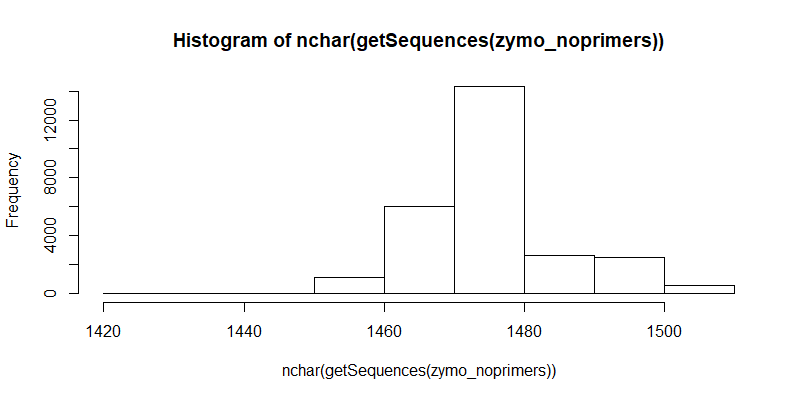
#Basic statistics of filtered contigs (full length 16S, primers trimmed)
summary(nchar(getSequences(zymo_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1426 1464 1472 1474 1474 1509
#Distribution of filtered contigs (full length 16S, primers trimmed)
zymo_noprimers <- file.path("zymo_noprimers.fastq")
hist(nchar(getSequences(zymo_noprimers)), 10)
Mapping to Provided References
Zymo replaced five strains in the Zymo standard with similar strains beginning with Lot ZRC190633. As has been noted elsewhere (Callahan Benjamin C., et al. “High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution”, Nucleic Acids Research, 47.18 (2019): e103), the exact strains used are not disclosed and the reference sequences provided by Zymo Research do not exactly correspond to the strains provided. There are two versions of the references: version 1 and version 2. In both versions, there are systematic nucleotide substitutions that cannot be explained by sequencing or contig assembly error. The references include whole genomes and 16S genes only. In version 1 of the references, the whole genome references are in multiple contigs and in the 16S gene references, only one 16S copy is given for each species. Version 2 of references aren’t correct either but is still more correct than version 1. The new version has complete whole genome references and all 16S gene copies are given for each species.
Concatenate all bacterial whole genome fasta files together and build a Bowtie2 index with version 2 of the whole genome references and map the filtered Zymo contigs.
cat *.fasta > zymo_bacteria_genome_ref.fasta
bowtie2-build zymo_bacteria_genome_ref.fasta zymo_bacteria_genome_ref
bowtie2 --very-sensitive -p 40 --un zymo.un.fastq -x zymo_bacteria_genome_ref/zymo_bacteria_genome_ref -q zymo_noprimers.fastq -S zymo.sam
27123 reads; of these:
27123 (100.00%) were unpaired; of these:
0 (0.00%) aligned 0 times
0 (0.00%) aligned exactly 1 time
27123 (100.00%) aligned >1 times
100.00% overall alignment rate
Many of the 16S genes are highly similar/identical and multiple valid alignments may occur. Even though the provided reference sequences are incorrect, given that the contigs are full-length 16S sequences and the contigs are accurate enough, all eight bacterial species are sufficiently phylogenetically distant that the multiple mappings occur within a single genome reference. It is the case generally that accurate full-length 16S sequences are able to discriminate between species (Johnson, Jethro S., et al. “Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis.” Nature communications 10.1 (2019): 1-11.). There are exceptions (eg. Mycobacterium genus) where inter-species 16S copies are so similar (~99.9%), that with even minor sequence errors, it may be difficult to unambiguously map the sequences to the correct references.
#Extract names of hits and count number of each unique hit
samtools view -F 4 zymo.sam | cut -f3 | sort | uniq -c
5692 BS.pilon.polished.v3.ST170922
2627 Enterococcus_faecalis_complete_genome
2962 Escherichia_coli_chromosome
3066 Lactobacillus_fermentum_complete_genome
4722 Listeria_monocytogenes_complete_genome
1151 Pseudomonas_aeruginosa_complete_genome
3032 Salmonella_enterica_complete_genome
3871 Staphylococcus_aureus_chromosome
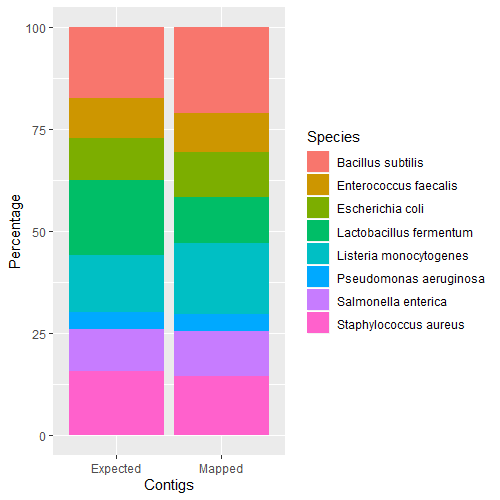
| Species | Expected contig % | Mapped contig % |
|---|---|---|
| Bacillus subtilis | 17.4 | 21.0 |
| Enterococcus faecalis | 9.9 | 9.7 |
| Escherichia coli | 10.1 | 10.9 |
| Lactobacillus fermentum | 18.4 | 11.3 |
| Listeria monocytogenes | 14.1 | 17.4 |
| Pseudomonas aeruginosa | 4.2 | 4.2 |
| Salmonella enterica | 10.4 | 11.2 |
| Staphylococcus aureus | 15.5 | 14.3 |
Species ratios are as expected.
#R
library(ggplot2)
zymo <- read.table("zymo_exp_vs_map.txt", header=TRUE, sep="\t")
ggplot(zymo, aes(fill=Species, y=Percentage, x=Contigs)) + geom_bar(position="stack", stat="identity")
Intergenomic 16S Gene Copies & Contig Accuracy
B. subtilis has the most 16S gene copies (ten) of the species in the Zymo standard so it can provide a fine-grain view of relative intergenomic 16S gene ratios. It also appears to be one of the correct references for the strain provided (no systematic mismatches between contig and reference) so it would allow for an unbiased assessment of LoopSeq contig accuracy.
Extract the full length 16S contigs that orginally mapped to B. subtilis.
samtools view zymo.sam | cut -f1,3 | awk -F "\t" '{ if($2 == "BS.pilon.polished.v3.ST170922") { print $1}}' > bsubtilis_contigs_list
seqtk subseq zymo_noprimers.fastq bsubtilis_contigs_list > bsubtilis_contigs.fastq
Sequences for each of the ten 16S genes is given in the reference fasta file. Multiple sequence alignment shows that there are five non-redundant copies . Copy #1, #4, #8, #9, #10 are identical. #2, #3 are identical. #5, #6, #7 are all unique. To prevent multi-mapping, build a bowtie2 index only using #1, #2, #5, #6, #7. Thus a mapping ratio of 5:2:1:1:1 is expected.
bowtie2 --very-sensitive -p 40 --un bsubtilis_contigs_trimmed.un.fastq -x zymo_bsubtilis_ref/Bacillus_subtilis_16S_nr -q bsubtilis_contigs.fastq -S bsubtilis.sam
5692 reads; of these:
5692 (100.00%) were unpaired; of these:
0 (0.00%) aligned 0 times
0 (0.00%) aligned exactly 1 time
5692 (100.00%) aligned >1 times
100.00% overall alignment rate #all mapped to 16S copies
#Extract reference hits
samtools view -F 4 bsubtilis.sam | cut -f3 | sort | uniq -c > bsubtilis_hits_counts
2466 Bacillus_subtilis_16S_1
1482 Bacillus_subtilis_16S_2
604 Bacillus_subtilis_16S_5
617 Bacillus_subtilis_16S_6
523 Bacillus_subtilis_16S_7
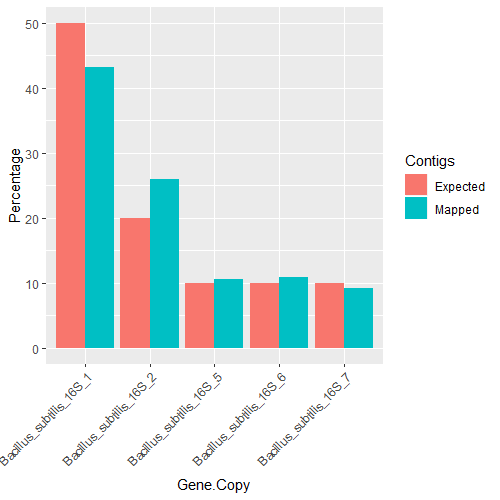
| 16S Gene Copy | Expected contig % | Mapped contig % |
|---|---|---|
| Bacillus_subtilis_16S_1 | 50 | 43.3 |
| Bacillus_subtilis_16S_2 | 20 | 26 |
| Bacillus_subtilis_16S_5 | 10 | 10.6 |
| Bacillus_subtilis_16S_6 | 10 | 10.8 |
| Bacillus_subtilis_16S_7 | 10 | 9.1 |
Intergenomic 16S gene ratios are as expected.
zymo_bsubtilis <- read.table("zymo_bsubtilis_16S_copies.txt", header=TRUE, sep="\t")
ggplot(zymo_bsubtilis, aes(fill=Contigs, y=Percentage, x=Gene.Copy)) +
geom_bar(position="dodge", stat="identity") +
theme(axis.text.x = element_text(angle = 45, hjust = 1))
Visually inspect the alignments to check if there is anything unusual about the alignments. Sort and index the SAM file and load in IGV.
samtools view -b -S bsubtilis.sam | samtools sort -o bsubtilis.sorted.bam - && samtools index bsubtilis.sorted.bam
Bacillus_subtilis_16S_1
Bacillus_subtilis_16S_2
Bacillus_subtilis_16S_5
Bacillus_subtilis_16S_6
Bacillus_subtilis_16S_7
Quantify the quality of the alignments in Alfred.
alfred qc -r zymo_bsubtilis_ref/Bacillus_subtilis_16S_nr.fasta -o alignment_qc.tsv.gz bsubtilis.sorted.bam
zgrep ^ME alignment_qc.tsv.gz | cut -f 2- | datamash transpose | column -t > zymo_bsubtilis_16S_alignment_qc.tsv
QC Summary
- MatchRate 0.999798
- MismatchRate 0.000201824
- DeletionRate 2.38704e-06
- InsertionRate 2.14834e-06
The error rates are extremely low considering these contigs have only been filtered to cover the full-length of the 16S gene. They have not been quality filtered, denoised or any other type of algorithmic error correction. Indels are negligible. Since consensus sequence accuracy is contigent on read depth, higher per base accuracy and a higher proportion of full-length 16S contigs can be expected with increased sequencing yield. But since there is a limited number of UMI barcodes per 16S fragment, there will be an upper limit to gains in accuracy.
Examine the distribution of mismatch errors in the contigs. Use a custom script to quantify the position and frequencies of variants.
samtools mpileup -B -L 1 -Q 0 -F 0 -f zymo_bsubtilis_ref/Bacillus_subtilis_16S_nr.fasta bsubtilis.sorted.bam > bsubtilis.pileup
pileup2SNVbaseCounts.pl -i bsubtilis.pileup -snvDepth 0 -indelDepth 0
The five non-redundant copies of the 16S gene are the same length, so they can be overlapped on the same plot. Only base mismatches are shown since there are practically no indels to visualize.
#R
bsubtilis_variants <- read.csv("zymo_bsubtilis_mpileup_baseCount.csv",header=TRUE)
ggplot(data=bsubtilis_variants,aes(x=bsubtilis_variants$Position,fill=Reference,y=bsubtilis_variants$Variant_Ratio)) + geom_bar(stat="identity")
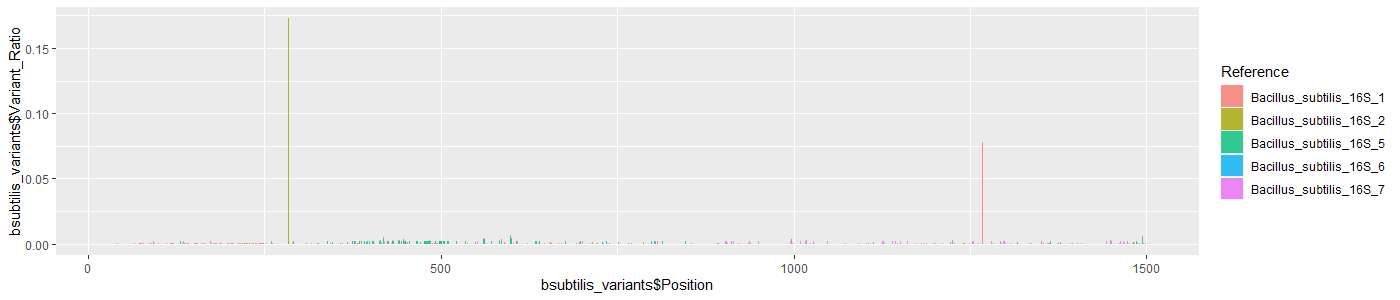
Mismatch errors are randomly distributed. The mismatches at the two peaks (gene copy #2 at pos: 285 [G->A] and gene copy #1 pos: 268 [G->A]) likely represent real variants since they are too high to be explained by sequencing or assembly error: the sequencing library consists of short randomly fragmented segments of the 16S gene. Consequently, the true mismatch error rate may be even lower: the bases in the two peaks combined accounts for 454/1691 total mismatch errors.
Comparison with Illumina Short Paired-End Reads
Short read 16S barcoding studies frequently sequence the V3V4 hypervariable region in 300bp paired-end mode. These paired-end reads are then filtered, trimmed and merged into ~440bp fragments which are then used for denoising and/or OTU clustering and classification. To make a fair comparison with LoopSeq contigs, the short reads are processed in a way as might be seen in a typical 16S barcoding study. The denoising step is omitted because it would potentially produce error-free reads; as would be the case if LoopSeq contigs were to undergo denoising. This would not permit a meaningful comparison of accuracies between the two methodologies.
The fastq files are too large to host on Github so have not been provided.
#Check how many pairs of reads in raw fastq file
echo $(zcat zymo-v3v4_R1.fastq.gz|wc -l)/4|bc
3278936
#Map against the PhiX reference to remove spike-in sequences
bowtie2 -p 24 --un-conc zymo-v3v4.fastq -x phiX/phiX -1 zymo-v3v4_R1.fastq.gz -2 zymo-v3v4_R2.fastq.gz -S zymo-v3v4_phiX.sam
#Filter out reads with low average quality and clip low quality bases in the tails
java -jar trimmomatic-0.36.jar PE zymo-v3v4.1.fastq.gz zymo-v3v4.2.fastq.gz zymo-3V4_clean_1.fastq.gz zymo-v3v4_unclean_1.fastq.gz zymo-v3v4_clean_2.fastq.gz zymo-v3v4_unclean_2.fastq.gz AVGQUAL:20
java -jar trimmomatic-0.36.jar PE zymo-v3v4_clean_1.fastq.gz zymo-v3v4_clean_2.fastq.gz zymo-v3v4_good_1.fastq.gz zymo-v3v4_bad_1.fastq.gz zymo-v3v4_good_2.fastq.gz zymo-v3v4_bad_2.fastq.gz TRAILING:20
#Merge reads
flash -t 24 -m 20 -M 250 -d merged -o zymo-v3v4 zymo-v3v4_good_1.fastq.gz zymo-v3v4_good_2.fastq.gz
#Trim V3V4 primers. 3’ primer sequence needs to be given in the reverse complement.
cutadapt -e 0.05 -g CCTACGGGNGGCWGCAG -a GGATTAGATACCCBDGTAGT -o zymo-v3v4.noprimers.fastq zymo-v3v4.extendedFrags.fastq --discard-untrimmed --no-indels
Check merged read distribution.
#R
zymo_v3V4_noprimers <- file.path("zymo-v3v4.noprimers.fastq")
summary(nchar(getSequences(zymo_v3V4_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
17.0 444.0 444.0 444.1 444.0 574.0
#Some merged reads are too short/long to be correct so these need to be filtered out
fastqFilter(zymo_v3V4_noprimers, "zymo-v3v4_noprimers_length.fastq", minLen=440, maxLen=450, verbose=TRUE, compress=FALSE)
Read in 2761523, output 2239559 (81.1%) filtered sequences.
#Map to Zymo whole genome bacteria references
bowtie2 --very-sensitive -p 40 --un zymo-v3v4.un.fastq -x zymo_bacteria_genome_ref/zymo_bacteria_genome_ref -q zymo-v3v4_noprimers_length.fastq -S zymo-v3v4.sam
2239559 reads; of these:
2239559 (100.00%) were unpaired; of these:
2051 (0.09%) aligned 0 times
51 (0.00%) aligned exactly 1 time
2237457 (99.91%) aligned >1 times
99.91% overall alignment rate
#Extract names of hits and count number of each unique hit
samtools view -F 4 zymo-v3v4.sam | cut -f3 | sort | uniq -c
348730 BS.pilon.polished.v3.ST170922
202215 Enterococcus_faecalis_complete_genome
266642 Escherichia_coli_chromosome
351894 Lactobacillus_fermentum_complete_genome
282619 Listeria_monocytogenes_complete_genome
177842 Pseudomonas_aeruginosa_complete_genome
268401 Salmonella_enterica_complete_genome
339165 Staphylococcus_aureus_chromosome
| Species | Expected % | Mapped % |
|---|---|---|
| Bacillus subtilis | 17.4 | 15.6 |
| Enterococcus faecalis | 9.9 | 9.0 |
| Escherichia coli | 10.1 | 11.9 |
| Lactobacillus fermentum | 18.4 | 15.7 |
| Listeria monocytogenes | 14.1 | 12.6 |
| Pseudomonas aeruginosa | 4.2 | 8.0 |
| Salmonella enterica | 10.4 | 12.0 |
| Staphylococcus aureus | 15.5 | 15.2 |
The species ratios are as expected. It does not appear that short read methodologies significanlty skew the relative abundances of the species examined.
To compare read accuracies with the LoopSeq contigs, extract the merged reads that mapped to B. subtilis and map the reads to the five non-redundant B. subtilis 16S gene copy references.
samtools view zymo-v3v4.sam | cut -f1,3 | awk -F "\t" '{ if($2 == "BS.pilon.polished.v3.ST170922") { print $1}}' > bsubtilis-v3v4_reads_list
seqtk subseq zymo-v3v4.noprimers.fastq bsubtilis-v3v4_reads_list > bsubtilis-v3v4_reads.fastq
bowtie2 --very-sensitive -p 40 --un bsubtilis-v3v4_reads.un.fastq -x zymo_bsubtilis_ref/Bacillus_subtilis_16S_nr -q bsubtilis-v3v4_reads.fastq -S bsubtilis-v3v4_reads.sam
348730 reads; of these:
348730 (100.00%) were unpaired; of these:
0 (0.00%) aligned 0 times
4 (0.00%) aligned exactly 1 time
348726 (100.00%) aligned >1 times
100.00% overall alignment rate
#Extract names of hits and count number of each unique hit
samtools view -F 4 bsubtilis-v3v4_reads.sam | cut -f3 | sort | uniq -c
78053 Bacillus_subtilis_16S_1
77557 Bacillus_subtilis_16S_2
38240 Bacillus_subtilis_16S_5
77371 Bacillus_subtilis_16S_6
77509 Bacillus_subtilis_16S_7
| 16S Gene Copy | Expected contig % | Mapped reads % |
|---|---|---|
| Bacillus_subtilis_16S_1 | 50 | 22.4 |
| Bacillus_subtilis_16S_2 | 20 | 22.3 |
| Bacillus_subtilis_16S_5 | 10 | 11.0 |
| Bacillus_subtilis_16S_6 | 10 | 22.2 |
| Bacillus_subtilis_16S_7 | 10 | 22.2 |
Many of the nucleotide differences between the five non-redundant B. subtilis 16S copies are not within the V3V4 region (pos: 348-792). Within the V3V4 region of B. subtilis, only 16S copy #5 is different (pos: 373 C->A) while copies #1, #2, #6 and #7 are all identical in the V3V4 region. Reads corresponding to these identical sequences would map equally well to any of the 16S copies so are randomly placed, resulting in the approximately equal ratios observed for copy #1, #2, #6 and #7. This shows that sequencing short hypervariable regions may not permit an accurate assessment of intergenomic 16S gene ratios. This limits the use of short reads in inferring bacterial subtypes which may be distinguished on the basis of their intergenomic 16S copies. In general, the use of short hypervariable regions (V1-V2, V1-V3, V4, etc) has limited utility in identifying prokaryotes to the species level (Johnson, Jethro S., et al. “Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis.” Nature Communications 10.1 (2019): 1-11.).
Quantify the quality of the alignments in Alfred.
samtools view -b -S bsubtilis-v3v4_reads.sam | samtools sort -o bsubtilis-v3v4_reads.sorted.bam - && samtools index bsubtilis-v3v4_reads.sorted.bam
alfred qc -r zymo_bsubtilis_ref/Bacillus_subtilis_16S_nr.fasta -o bsubtilis-v3v4_alignment_qc.tsv.gz bsubtilis-v3v4_reads.sorted.bam
zgrep ^ME bsubtilis-v3v4_alignment_qc.tsv.gz | cut -f 2- | datamash transpose | column -t > zymo_bsubtilis-v3v4_alignment_qc.tsv
QC Summary
- MatchRate 0.992513
- MismatchRate 0.00748692
- DeletionRate 8.36867e-05
- InsertionRate 3.11562e-05
The mismatch, insertion and deletion error rates for short reads are at least an order of magnitude larger than the LoopSeq contigs. Optimized experimental protocols and stricter quality filtering criteria may result in reads with even lower mean error rates. There are also denoising algorithms that can correct PCR and sequencing errors, but such corrected reads would not accurately reflect the underlying error profile. Conversely, not performaing any quality filtering would result in reads with a higher error rate, thus not meeting basic quality standards for analysis. The filtering utilized above simply represents a typical pre-processing workflow which allows us to make a fair comparison with LoopSeq contigs which have only been filtered to full-length.
Mapping to NCBI RefSeq References
The eight bacterial species in the Zymo mock community are sufficiently phylogenetically distant that if given reads/contigs are long and accurate enough, reads/contigs are unlikely to me errorneously mapped to the wrong species. However, it is not always the case that species within a sample are known and/or reference sequences for the species are available. It it more often the case that samples with unknown composition are compared to a more general database. To determine if full-length 16S sequences can be used to accurately identify the correct species using a general database, the full-length LoopSeq 16S Zymo contigs were mapped to all bacterial genomes in the NCBI RefSeq CG (complete genomes) database as of October 2019. The database contains genomes of the same species as those in the Zymo sample (although it is unclear if the strains are the same since this information is lacking). There are also many genomes of closely related species (species from the same genus) that could potentially counfound a direct mapping approach to species identification.
Bacterial genomes were download from NCBI RefSeq using Kraken2. A Bowtie2 index was built (this takes some time) and full-length LoopSeq 16S contigs were mapped to the database.
bowtie2 --very-sensitive -p 40 --un zymo_refseq.un.fastq -x bt2_refseq_bacteria_genomes/refseq_bacteria -q zymo_noprimers.fastq -S zymo_refseq.sam
27123 reads; of these:
27123 (100.00%) were unpaired; of these:
0 (0.00%) aligned 0 times
1 (0.00%) aligned exactly 1 time
27122 (100.00%) aligned >1 times
100.00% overall alignment rate
#Extract reference hits
samtools view -F 4 zymo_refseq.sam | cut -f3 > zymo_refseq_hits
The bacterial references and taxonomy files were downloaded with Kraken2. The header does not contain the original species names. It does have taxids which can be converted and reformatted using taxonkit.
#Cut out the taxids from the header and convert to a 7-rank taxonomy
cat zymo_refseq_hits | cut -f2 -d"|" \
| taxonkit lineage --data-dir Kraken2/standard_DB/taxonomy \
| taxonkit reformat --data-dir Kraken2/standard_DB/taxonomy \
| cut -f3 | sort | uniq -c
2 Bacteria;Firmicutes;Bacilli;Bacillales;Bacillaceae;Bacillus;Bacillus halotolerans
1 Bacteria;Firmicutes;Bacilli;Bacillales;Bacillaceae;Bacillus;Bacillus sp. JS
1 Bacteria;Firmicutes;Bacilli;Bacillales;Bacillaceae;Bacillus;Bacillus sp. SJ-10
5689 Bacteria;Firmicutes;Bacilli;Bacillales;Bacillaceae;Bacillus;Bacillus subtilis
4722 Bacteria;Firmicutes;Bacilli;Bacillales;Listeriaceae;Listeria;Listeria monocytogenes
81 Bacteria;Firmicutes;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus argenteus
3789 Bacteria;Firmicutes;Bacilli;Bacillales;Staphylococcaceae;Staphylococcus;Staphylococcus aureus
2627 Bacteria;Firmicutes;Bacilli;Lactobacillales;Enterococcaceae;Enterococcus;Enterococcus faecalis
3066 Bacteria;Firmicutes;Bacilli;Lactobacillales;Lactobacillaceae;Lactobacillus;Lactobacillus fermentum
2952 Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Enterobacteriaceae;Escherichia;Escherichia coli
1 Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Enterobacteriaceae;Klebsiella;Klebsiella aerogenes
3032 Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Enterobacteriaceae;Salmonella;Salmonella enterica
7 Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Enterobacteriaceae;Shigella;Shigella dysenteriae
2 Bacteria;Proteobacteria;Gammaproteobacteria;Enterobacterales;Enterobacteriaceae;Shigella;Shigella flexneri
1142 Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Pseudomonadaceae;Pseudomonas;Pseudomonas aeruginosa
7 Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Pseudomonadaceae;Pseudomonas;Pseudomonas fluorescens
2 Bacteria;Proteobacteria;Gammaproteobacteria;Pseudomonadales;Pseudomonadaceae;Pseudomonas;Pseudomonas sp. AK6U
| Species | Expected % | Mapped % |
|---|---|---|
| Bacillus subtilis | 17.4 | 21.0 |
| Enterococcus faecalis | 9.9 | 9.7 |
| Escherichia coli | 10.1 | 10.9 |
| Lactobacillus fermentum | 18.4 | 11.3 |
| Listeria monocytogenes | 14.1 | 17.4 |
| Pseudomonas aeruginosa | 4.2 | 4.2 |
| Salmonella enterica | 10.4 | 11.2 |
| Staphylococcus aureus | 15.5 | 14.0 |
| Other species | 0 | 0.4 |
#R
library(ggplot2)
zymo_refseq <- read.table("zymo_refseq_exp_vs_map.txt", header=TRUE, sep="\t")
ggplot(zymo_refseq, aes(fill=Species, y=Percentage, x=Contigs, group=customOrder)) + geom_bar(position="stack", stat="identity")
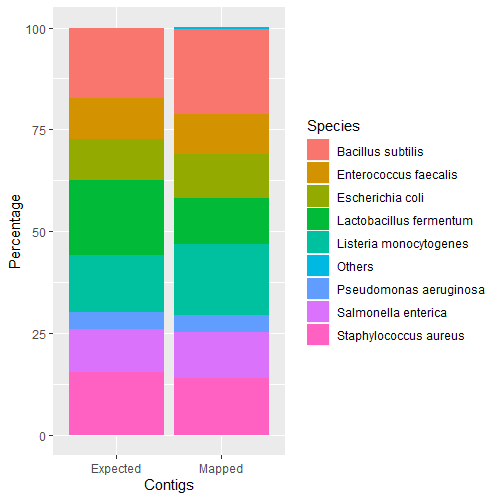
Only 0.4% of contigs were mapped to incorrect species. With the exception of a single contig that mapped to a different genus (Klebsiella), the incorrect mappings were consistent with the genuses in the Zymo sample. This demonstrates that it is possible to accurately classify bacteria to the species level using full-length LoopSeq 16S contigs against a general whole genome database. This also demonstrates that even minor differences may produce incorrect species mappings.
Classification with LCA Classifier
Although the direct mapping approach may provide higher classification precision when references are available, it is frequently the case that exact references are not availble. One may nontheless still be interested in classifying the sequence to higher taxonomic levels (genus, family etc.). For such cases, lowest common ancestor (LCA) classifiers may be utilized. Commonly used 16S classification tools include mothur and QIIME used in conjunction with the SILVA, GreenGenes or RDP databases. The databases may contain 16S sequences from many representative species but the full intergenomic 16S diversity is usually not represented. Thus, it may be more advantageous to use whole genome references (which contain all the 16S copies) when they are available. The obvious disadvantage is that fewer species are represented as compared to 16S databases.
Kraken2 is a LCA classifer that searches for exact alignment of k-mers against an index built from whole genome references. The database used is the standard Kraken2 database (NCBI RefSeq CG: bacteria, archaea, virus, human, common cloning vectors).
kraken2 --db Kraken2/standard_DB --threads 24 --out zymo.kraken --report zymo.kreport zymo_noprimers.fastq
The result columns are interpreted as follows:
- Percentage of fragments covered by the clade rooted at this taxon
- Number of fragments covered by the clade rooted at this taxon
- Number of fragments assigned directly to this taxon
- A rank code, indicating (U)nclassified, (R)oot, (D)omain, (K)ingdom, (P)hylum, (C)lass, (O)rder, (F)amily, (G)enus, or (S)pecies. Taxa that are not at any of these 10 ranks have a rank code that is formed by using the rank code of the closest ancestor rank with a number indicating the distance from that rank. E.g., “G2” is a rank code indicating a taxon is between genus and species and the grandparent taxon is at the genus rank.
- NCBI taxonomic ID number
- Indented scientific name
100.00 27123 0 R 1 root
100.00 27123 0 R1 131567 cellular organisms
100.00 27123 0 D 2 Bacteria
73.66 19978 0 D1 1783272 Terrabacteria group
73.66 19978 0 P 1239 Firmicutes
73.66 19978 0 C 91061 Bacilli
52.66 14284 0 O 1385 Bacillales
20.98 5691 0 F 186817 Bacillaceae
20.98 5691 21 G 1386 Bacillus
20.86 5658 3877 G1 653685 Bacillus subtilis group
6.51 1767 1759 S 1423 Bacillus subtilis
0.03 7 4 S1 135461 Bacillus subtilis subsp. subtilis
0.00 1 1 S2 224308 Bacillus subtilis subsp. subtilis str. 168
0.00 1 1 S2 1052588 Bacillus subtilis subsp. subtilis str. RO-NN-1
0.00 1 1 S2 1302650 Bacillus subtilis subsp. subtilis str. BAB-1
0.00 1 0 S1 96241 Bacillus subtilis subsp. spizizenii
0.00 1 1 S2 1052585 Bacillus subtilis subsp. spizizenii TU-B-10
0.04 11 0 G2 1938374 Bacillus amyloliquefaciens group
0.03 8 8 S 492670 Bacillus velezensis
0.01 3 2 S 1390 Bacillus amyloliquefaciens
0.00 1 1 S1 1034836 Bacillus amyloliquefaciens XH7
0.01 2 2 S 1402 Bacillus licheniformis
0.00 1 1 S 119858 Bacillus sonorensis
0.02 5 1 G1 86661 Bacillus cereus group
0.01 3 1 S 1396 Bacillus cereus
0.00 1 1 S1 1454382 Bacillus cereus D17
0.00 1 0 S1 1179100 Bacillus cereus biovar anthracis
0.00 1 1 S2 637380 Bacillus cereus biovar anthracis str. CI
0.00 1 0 S 1392 Bacillus anthracis
0.00 1 1 S1 673518 Bacillus anthracis str. A16R
0.01 3 3 S 152268 Bacillus litoralis
0.01 2 0 S 1404 Bacillus megaterium
0.01 2 2 S1 592022 Bacillus megaterium DSM 319
0.01 2 0 S 665099 Bacillus oceanisediminis
0.01 2 2 S1 1196031 Bacillus oceanisediminis 2691
17.41 4722 0 F 186820 Listeriaceae
17.41 4722 0 G 1637 Listeria
17.41 4722 4722 S 1639 Listeria monocytogenes
14.27 3871 0 F 90964 Staphylococcaceae
14.27 3871 2725 G 1279 Staphylococcus
4.14 1122 1115 S 1280 Staphylococcus aureus
0.03 7 1 S1 46170 Staphylococcus aureus subsp. aureus
0.02 5 5 S2 985006 Staphylococcus aureus subsp. aureus LGA251
0.00 1 1 S2 548473 Staphylococcus aureus subsp. aureus TCH60
0.01 4 4 S 1295 Staphylococcus schleiferi
0.01 3 3 S 29379 Staphylococcus auricularis
0.01 3 3 S 28035 Staphylococcus lugdunensis
0.01 2 2 S 1282 Staphylococcus epidermidis
0.01 2 2 S 29384 Staphylococcus kloosii
0.00 1 1 S 70258 Staphylococcus piscifermentans
0.00 1 1 S 70255 Staphylococcus condimenti
0.00 1 1 S 246432 Staphylococcus equorum
0.00 1 0 S 29388 Staphylococcus capitis
0.00 1 1 S1 72758 Staphylococcus capitis subsp. capitis
0.00 1 1 S 29385 Staphylococcus saprophyticus
0.00 1 1 S 283734 Staphylococcus pseudintermedius
0.00 1 1 S 308354 Staphylococcus simiae
0.00 1 1 S 1286 Staphylococcus simulans
0.00 1 1 S 1283 Staphylococcus haemolyticus
0.00 1 1 S 2044912 Staphylococcus sp. SDB 2975
20.99 5694 0 O 186826 Lactobacillales
11.30 3066 0 F 33958 Lactobacillaceae
11.30 3066 0 G 1578 Lactobacillus
11.30 3066 3064 S 1613 Lactobacillus fermentum
0.01 2 2 S1 767453 Lactobacillus fermentum F-6
9.69 2628 0 F 81852 Enterococcaceae
9.69 2628 0 G 1350 Enterococcus
9.69 2628 2628 S 1351 Enterococcus faecalis
26.34 7145 0 P 1224 Proteobacteria
26.34 7145 0 C 1236 Gammaproteobacteria
22.10 5994 1 O 91347 Enterobacterales
22.10 5993 2038 F 543 Enterobacteriaceae
11.19 3034 0 G 590 Salmonella
11.19 3034 3013 S 28901 Salmonella enterica
0.08 21 9 S1 59201 Salmonella enterica subsp. enterica
0.01 3 1 S2 149539 Salmonella enterica subsp. enterica serovar Enteritidis
0.01 2 2 S3 1244111 Salmonella enterica subsp. enterica serovar Enteritidis str. EC20110357
0.01 2 0 S2 57046 Salmonella enterica subsp. enterica serovar Paratyphi C
0.01 2 2 S3 476213 Salmonella enterica subsp. enterica serovar Paratyphi C str. RKS4594
0.00 1 1 S2 90370 Salmonella enterica subsp. enterica serovar Typhi
0.00 1 0 S2 90371 Salmonella enterica subsp. enterica serovar Typhimurium
0.00 1 1 S3 1008297 Salmonella enterica subsp. enterica serovar Typhimurium str. 798
0.00 1 1 S2 98360 Salmonella enterica subsp. enterica serovar Dublin
0.00 1 0 S2 108619 Salmonella enterica subsp. enterica serovar Newport
0.00 1 1 S3 1454618 Salmonella enterica subsp. enterica serovar Newport str. USDA-ARS-USMARC-1925
0.00 1 1 S2 605 Salmonella enterica subsp. enterica serovar Pullorum
0.00 1 0 S2 58712 Salmonella enterica subsp. enterica serovar Anatum
0.00 1 1 S3 1454592 Salmonella enterica subsp. enterica serovar Anatum str. CDC 06-0532
0.00 1 1 S2 29474 Salmonella enterica subsp. enterica serovar California
3.33 903 0 G 561 Escherichia
3.33 902 899 S 562 Escherichia coli
0.01 2 0 S1 83333 Escherichia coli K-12
0.01 2 2 S2 1110693 Escherichia coli str. K-12 substr. MDS42
0.00 1 1 S1 2048777 Escherichia coli O15:H11
0.00 1 1 S 1499973 Escherichia marmotae
0.04 10 3 G 620 Shigella
0.03 7 3 S 623 Shigella flexneri
0.01 3 0 S1 424718 Shigella flexneri 5a
0.01 3 3 S2 1086030 Shigella flexneri 5a str. M90T
0.00 1 1 S1 42897 Shigella flexneri 2a
0.01 4 0 G 570 Klebsiella
0.01 4 4 S 573 Klebsiella pneumoniae
0.01 3 0 G 547 Enterobacter
0.01 2 0 G1 354276 Enterobacter cloacae complex
0.00 1 1 S 550 Enterobacter cloacae
0.00 1 1 S 1915310 Enterobacter cloacae complex sp. ECNIH7
0.00 1 1 S 881260 Enterobacter bugandensis
0.00 1 0 F1 191675 unclassified Enterobacteriaceae
0.00 1 0 F2 84563 ant, tsetse, mealybug, aphid, etc. endosymbionts
0.00 1 0 F3 146507 aphid secondary symbionts
0.00 1 1 S 1199245 secondary endosymbiont of Ctenarytaina eucalypti
4.24 1151 0 O 72274 Pseudomonadales
4.24 1151 0 F 135621 Pseudomonadaceae
4.24 1151 1143 G 286 Pseudomonas
0.01 2 0 S 101564 Pseudomonas alcaliphila
0.01 2 2 S1 741155 Pseudomonas alcaliphila JAB1
0.01 2 0 G1 136842 Pseudomonas chlororaphis group
0.01 2 2 S 296 Pseudomonas fragi
0.00 1 1 S 1981174 Pseudomonas sp. M30-35
0.00 1 1 S 1302376 Candidatus Pseudomonas adelgestsugas
0.00 1 0 G1 136841 Pseudomonas aeruginosa group
0.00 1 1 S 287 Pseudomonas aeruginosa
0.00 1 0 G1 136843 Pseudomonas fluorescens group
0.00 1 1 S 47883 Pseudomonas synxantha
Most contigs were correctly assigned to the species level. The exception was P. aureginosa, for which most contigs could only be assigned to the genus level. However, the percentage of contigs (4.24%) that were classified to the Pseudomonas genus does conform to expectations. This lack of species resolution for P. aureginosa is due to characteristics of the Pseudomonas genus in general and not a particular shortcoming of LoopSeq contigs. A previous study (Gomila, Margarita, et al. “Phylogenomics and systematics in Pseudomonas.” Frontiers in Microbiology 6 (2015): 214.) found that:
In conclusion, because the resolution of the 16S rRNA tree was not sufficient to differentiate 63 genomes from other closely related Pseudomonas species, the classification of these bacteria should follow the phylogeny of the housekeeping genes until the whole genome sequence of the type strains of all Pseudomonas species is known.
Using Bracken, the companion tool to Kraken that redistributes all the classified reads to a single taxonomic level, it is apparent that at the genus level, the ratios of all eight species are as expected.
bracken -d Kraken2/standard_DB -i zymo.kreport -o zymo_genus.bracken -r 1500 -l G -t 1
| Genus | Expected % | Classified % |
|---|---|---|
| Bacillus | 17.4 | 21.0 |
| Enterococcus | 9.9 | 9.7 |
| Escherichia | 10.1 | 10.0 |
| Lactobacillus | 18.4 | 11.3 |
| Listeria | 14.1 | 17.4 |
| Pseudomonas | 4.2 | 4.2 |
| Salmonella | 10.4 | 11.9 |
| Staphylococcus | 15.5 | 14.3 |
| Other species | 0 | 0.3 |
Redistributing contigs to the species level results in a higher proportion of false positives. Of the expected 4.2% P. aureginosa species, approximately 3/4 were classied to other Pseudomonas species.
bracken -d Kraken2/standard_DB -i zymo.kreport -o zymo_species.bracken -r 1500 -l S -t 1
| Species | Expected % | Classified % |
|---|---|---|
| Bacillus subtilis | 17.4 | 19.0 |
| Enterococcus faecalis | 9.9 | 9.7 |
| Escherichia coli | 10.1 | 10.0 |
| Lactobacillus fermentum | 18.4 | 11.3 |
| Listeria monocytogenes | 14.1 | 17.4 |
| Salmonella enterica | 10.4 | 11.9 |
| Staphylococcus aureus | 15.5 | 13.8 |
| Pseudomonas aeruginosa | 4.2 | 1.0 |
| Other Pseudomonas spp. | 0 | 3.3 |
| Other species | 0 | 2.6 |
ATCC Mock Human Communities Analysis
The two ATCC mock human communities were analyzed using two methods:
Data Preparation
Mock Community Description
Although the Zymo mock community is widely used and is arguably the common standard in microbiome studies, many of the references that are provided do not match the strains in the sample. Zymo Research also does not offer the extensive supporting materials that the American Type Culture Association (ATCC) offers through their mock community samples (BEI Resources, USA). Many of the species in the ATCC mock communities have exact strain genome references along with assembly details and genome annotations. Where exact reference strains are not provided, strains names allows researchers to look for identical or similar references in common genome repositories (eg. NCBI RefSeq CG database).
Two ATCC mock communities representing human-derived samples were analyzed. These two mock communities demonstrates the utility of applying LoopSeq for human site-specific studies. The provided exact strain names also allows for a more fine-grained assessment of the level of taxonomic resolution that can be achieved through accurate full-length 16S sequences. Two mock communities were analyzed: ATCC-gut and ATCC-oral. Six out of the 12 genome references are provided by the ATCC for ATCC-gut, while the remaining six have equivalent NCBI RefSeq assemblies. All six of genome references are provided by the ATCC for ATCC-oral.
Gut Microbiome Genomic Mix (MSA-1006) (“ATCC-gut”)
| Species | Strain | Genomic DNA % | ATCC genome reference | Reference | RefSeq assembly | RefSeq assembly strain | 16S copy information | 16S copies |
|---|---|---|---|---|---|---|---|---|
| Bacteriodes fragilis | ATCC 25285 | 8.3 | No | link | Yes | NCTC 9343 (ATCC 25285) | Yes | 6 |
| Bacteroides vulgatus | ATCC 8482 | 8.3 | No | link | Yes | ATCC 8482 | Yes | 7 |
| Clostridioides difficile | ATCC 9689 | 8.3 | No | link | Yes | ATCC 9689 | Yes | 12 |
| Enterobacter cloacae | ATCC 13047 | 8.3 | No | link | Yes | ATCC 13047 | link | 8 |
| Escherichia coli | ATCC 700926 | 8.3 | No | link | Yes | MG1655 (ATCC 700926) | link | 7 |
| Salmonella enterica subsp. enterica | ATCC 9150 | 8.3 | No | link | Yes | ATCC 9150 | Yes | 7 |
| Bifidobacterium adolescentis | ATCC 15703 | 8.3 | Yes | link | 5 | |||
| Enterococcus faecalis | ATCC 700802 | 8.3 | Yes | link | 4 | |||
| Lactobacillus plantarum | ATCC BAA-793 | 8.3 | Yes | link | 5 | |||
| Helicobacter pylori | ATCC 700392 | 8.3 | Yes | link | 2 | |||
| Yersinia enterocolitica | ATCC 27729 | 8.3 | Yes | link | 7 | |||
| Fusobacterium nucleatum subsp. nucleatum | ATCC 25586 | 8.3 | Yes | link | 5 |
Oral Microbiome Genomic Mix (MSA-1004) (“ATCC-oral”)
| Species | Strain | Genomic DNA % | ATCC genome reference | Reference | 16S copies |
|---|---|---|---|---|---|
| Schaalia odontolytica | ATCC 17982 | 16.7 | Yes | link | 3 |
| Prevotella melaninogenica | ATCC 25845 | 16.7 | Yes | link | 4 |
| Fusobacterium nucleatum subsp. nucleatum | ATCC 25586 | 16.7 | Yes | link | 5 |
| Streptococcus mitis | ATCC 49456 | 16.7 | Yes | link | 4 |
| Veillonella parvula | ATCC 17745 | 16.7 | Yes | link | 4 |
| Haemophilus parainfluenzae | ATCC 33392 | 16.7 | Yes | link | 6 |
LoopSeq 16S Contig QC & Filtering
ATCC-gut
Load fastq file containing raw contigs as downloaded from the Loop Genomics cloud platform.
#R
library(dada2)
library(ShortRead)
F_prim <- "AGAGTTTGATCMTGGCTCAG" #forward primer
R_prim <- "TACCTTGTTACGACTT" #reverse primer
atcc_gut <- file.path("atcc_gut.fastq")
#Basic statistics of raw contigs
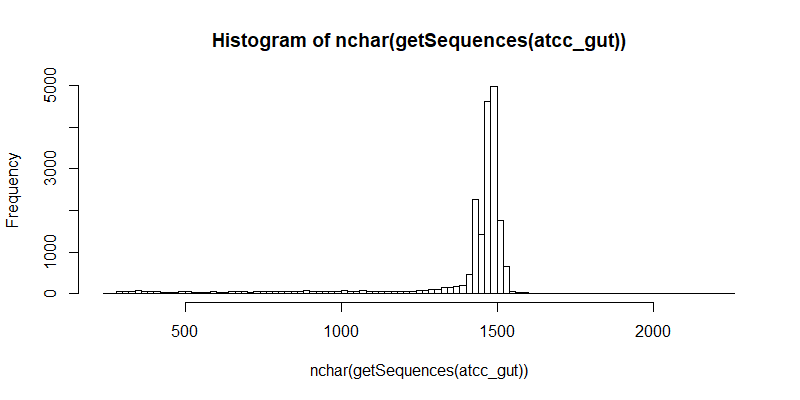
summary(nchar(getSequences(atcc_gut)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
259 1436 1462 1376 1489 2242
#Distribution of raw contigs
hist(nchar(getSequences(atcc_gut)), 100)
#Remove forward and reverse primers. This step also ensures that the contigs that remain after filtering are the full length 16S gene.
removePrimers(atcc_gut, "atcc_gut_noprimers.fastq", primer.fwd=F_prim, primer.rev=rc(R_prim), compress=FALSE, max.mismatch=2, allow.indels=FALSE, verbose=TRUE)
Multiple matches to the primer(s) in some sequences. Using the longest possible match.
Multiple matches to the primer(s) in some reverse-complement sequences. Using the longest possible match.
86 sequences out of 20027 are being reverse-complemented.
Read in 20027, output 9087 (45.4%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed)
atcc_gut_noprimers <- file.path("atcc_gut_noprimers.fastq")
summary(nchar(getSequences(atcc_gut_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1416 1426 1451 1451 1464 1585
#Distribution of filtered contigs (full length 16S, primers trimmed)
hist(nchar(getSequences(atcc_gut_noprimers)), 10)
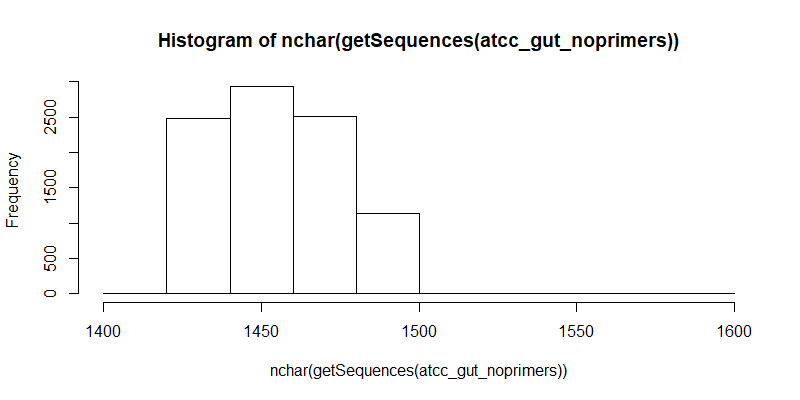
ATCC-oral
Load fastq file containing raw contigs as downloaded from the Loop Genomics cloud platform.
#R
library(dada2)
library(ShortRead)
F_prim <- "AGAGTTTGATCMTGGCTCAG" #forward primer
R_prim <- "TACCTTGTTACGACTT" #reverse primer
atcc_oral <- file.path("atcc_oral.fastq")
#Basic statistics of raw contigs
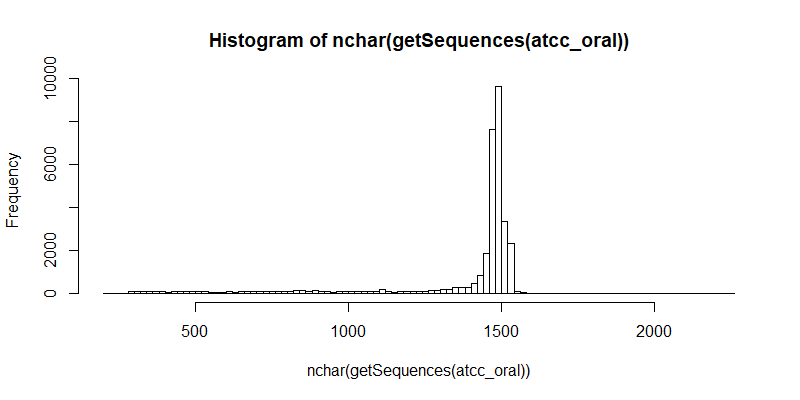
summary(nchar(getSequences(atcc_oral)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
205 1444 1478 1372 1498 2255
#Distribution of raw contigs
hist(nchar(getSequences(atcc_oral)), 100)
#Remove forward and reverse primers. This step also ensures that the contigs that remain after filtering are the full length 16S gene.
removePrimers(atcc_oral, "atcc_oral_noprimers.fastq"), primer.fwd=F_prim, primer.rev=rc(R_prim), compress=FALSE, max.mismatch=2, allow.indels=FALSE, verbose=TRUE)
Multiple matches to the primer(s) in some sequences. Using the longest possible match.
193 sequences out of 33222 are being reverse-complemented.
Read in 33222, output 16392 (49.3%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed)
atcc_oral_noprimers <- file.path("atcc_oral_noprimers.fastq")
summary(nchar(getSequences(atcc_oral_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
300 1442 1462 1460 1469 2219
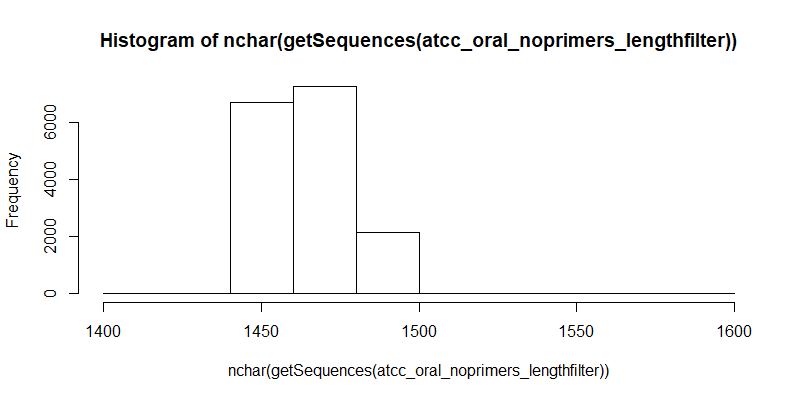
#The max contig length after primer trimming is 2219 bp. 16S genes shouldn't be that long. There are also contigs that are too short. These may represent experimental or assembly errors. These are filtered out.
filterAndTrim(atcc_oral_noprimers, "atcc_oral_noprimers_lengthfilter.fastq", minLen=1400, maxLen=1600, verbose=TRUE, compress=FALSE)
Read in 16392, output 16173 (98.7%) filtered sequences.
atcc_oral_noprimers_lengthfilter <- file.path("atcc_oral_noprimers_lengthfilter.fastq")
summary(nchar(getSequences(atcc_oral_noprimers_lengthfilter)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1409 1442 1462 1460 1469 1595
#Distribution of filtered contigs (full length 16S, primers trimmed, length filtered)
hist(nchar(getSequences(atcc_oral_noprimers_lengthfilter)), 10)
Mapping to Whole Genome References
ATCC-gut
Concatenate all bacterial whole genome fasta files together and build a Bowtie2 index with the whole genome references and map the filtered ATCC-gut contigs.
bowtie2 --very-sensitive -p 40 --un atcc_gut.un.fastq -x atcc_gut_ref/atcc_gut_ref -q atcc_gut_noprimers.fastq -S atcc_gut.sam
9087 reads; of these:
9087 (100.00%) were unpaired; of these:
3 (0.03%) aligned 0 times
0 (0.00%) aligned exactly 1 time
9084 (99.97%) aligned >1 times
99.97% overall alignment rate
#Extract names of hits and count number of each unique hit
samtools view -F 4 atcc_gut.sam | cut -f3 | sort | uniq -c
1067 Bacteroides_fragilis_NCTC_9343
968 Bacteroides_vulgatus_ATCC_8482
20 bifidobacterium_adolescentis_atcc_15703_contig1
11 bifidobacterium_adolescentis_atcc_15703_contig2
1966 Clostridioides_difficile_ATCC_9689
692 Enterobacter_cloacae_ATCC_13047
582 enterococcus_faecalis_atcc_700802_contig1
735 Escherichia_coli_MG1655
894 fusobacterium_nucleatum_subsp_nucleatum_atcc_25586
531 helicobacter_pylori_atcc_700392
556 lactobacillus_plantarum_atcc_baa-793_contig1
391 Salmonella_enterica_ATCC_9150
671 yersinia_enterocolitica_atcc_27729_contig1
| Species | Expected contig % | Mapped contig % |
|---|---|---|
| Bacteriodes fragilis | 8.0 | 11.7 |
| Bacteroides vulgatus | 9.3 | 10.7 |
| Clostridioides difficile | 16.0 | 21.6 |
| Enterobacter cloacae | 10.7 | 7.6 |
| Escherichia coli | 9.3 | 8.1 |
| Salmonella enterica subsp. enterica | 9.3 | 4.3 |
| Bifidobacterium adolescentis | 6.7 | 0.3 |
| Enterococcus faecalis | 5.3 | 6.4 |
| Lactobacillus plantarum | 6.7 | 6.1 |
| Helicobacter pylori | 2.7 | 5.8 |
| Yersinia enterocolitica | 9.3 | 7.4 |
| Fusobacterium nucleatum subsp. nucleatum | 6.7 | 9.8 |
The proportion of species 16S ratios are converted from 16S copy numbers. All 12 species are observed in approximately the expected ratios, except B. adolescentis which is much lower than expected.
ATCC-oral
Concatenate all bacterial whole genome fasta files together and build a Bowtie2 index with the whole genome references and map the filtered ATCC-oral contigs.
bowtie2 --very-sensitive -p 40 --un atcc_oral.un.fastq -x atcc_oral_ref/atcc_oral_ref -q atcc_oral_noprimers_lengthfilter.fastq -S atcc_oral.sam
16173 reads; of these:
16173 (100.00%) were unpaired; of these:
1 (0.01%) aligned 0 times
0 (0.00%) aligned exactly 1 time
16172 (99.99%) aligned >1 times
99.99% overall alignment rate
#Extract names of hits and count number of each unique hit
samtools view -F 4 atcc_oral.sam | cut -f3 | sort | uniq -c
4542 fusobacterium_nucleatum_subsp_nucleatum_atcc_25586
4144 haemophilus_parainfluenzae_atcc_33392_contig1
1083 prevotella_melaninogenica_atcc_25845_contig1
1129 prevotella_melaninogenica_atcc_25845_contig2
623 schaalia_odontolytica_atcc_17982
2485 streptococcus_mitis_atcc_49456
2166 veillonella_parvula_atcc_17745
| Species | Expected contig % | Mapped contig % |
|---|---|---|
| Schaalia odontolytica | 11.5 | 3.9 |
| Prevotella melaninogenica | 15.4 | 13.7 |
| Fusobacterium nucleatum subsp. nucleatum | 19.2 | 28.1 |
| Streptococcus mitis | 15.4 | 15.4 |
| Veillonella parvula | 15.4 | 13.4 |
| Haemophilus parainfluenzae | 23.1 | 25.6 |
Strain-level Detection Using LCA Classifier
ATCC-gut
kraken2 --db Kraken2/standard_DB --threads 24 --out atcc_gut.kraken --report atcc_gut.kreport atcc_gut_noprimers.fastq
Loading database information... done.
9087 sequences (13.19 Mbp) processed in 0.384s (1418.5 Kseq/m, 2058.48 Mbp/m).
9087 sequences classified (100.00%)
0 sequences unclassified (0.00%)
100.00 9087 0 R 1 root
100.00 9087 0 R1 131567 cellular organisms
100.00 9087 0 D 2 Bacteria
34.52 3137 0 D1 1783272 Terrabacteria group
34.18 3106 0 P 1239 Firmicutes
21.65 1967 0 C 186801 Clostridia
21.65 1967 0 O 186802 Clostridiales
21.64 1966 0 F 186804 Peptostreptococcaceae
21.64 1966 0 G 1870884 Clostridioides
21.64 1966 1965 S 1496 Clostridioides difficile
0.01 1 1 S1 699037 Clostridioides difficile M68
0.01 1 0 F 31979 Clostridiaceae
0.01 1 0 G 1485 Clostridium
0.01 1 1 S 1502 Clostridium perfringens
12.52 1138 0 C 91061 Bacilli
12.52 1138 0 O 186826 Lactobacillales
6.40 582 0 F 81852 Enterococcaceae
6.40 582 0 G 1350 Enterococcus
6.40 582 582 S 1351 Enterococcus faecalis
6.12 556 0 F 33958 Lactobacillaceae
6.12 556 229 G 1578 Lactobacillus
3.57 324 324 S 1590 Lactobacillus plantarum
0.01 1 1 S 1613 Lactobacillus fermentum
0.01 1 0 S 109790 Lactobacillus jensenii
0.01 1 1 S1 525329 Lactobacillus jensenii JV-V16
0.01 1 1 S 1610 Lactobacillus coryniformis
0.01 1 0 C 909932 Negativicutes
0.01 1 0 O 1843489 Veillonellales
0.01 1 0 F 31977 Veillonellaceae
0.01 1 0 G 29465 Veillonella
0.01 1 1 S 29466 Veillonella parvula
0.34 31 0 P 201174 Actinobacteria
0.34 31 0 C 1760 Actinobacteria
0.34 31 0 O 85004 Bifidobacteriales
0.34 31 0 F 31953 Bifidobacteriaceae
0.34 31 0 G 1678 Bifidobacterium
0.34 31 29 S 1680 Bifidobacterium adolescentis
0.02 2 2 S1 367928 Bifidobacterium adolescentis ATCC 15703
33.23 3020 0 P 1224 Proteobacteria
27.39 2489 0 C 1236 Gammaproteobacteria
27.39 2489 0 O 91347 Enterobacterales
20.00 1817 1503 F 543 Enterobacteriaceae
2.45 223 0 G 561 Escherichia
2.45 223 222 S 562 Escherichia coli
0.01 1 0 S1 1072458 Escherichia coli O7:K1
0.01 1 1 S2 1072459 Escherichia coli O7:K1 str. CE10
0.77 70 0 G 590 Salmonella
0.77 70 56 S 28901 Salmonella enterica
0.14 13 13 S1 59201 Salmonella enterica subsp. enterica
0.01 1 0 S1 59202 Salmonella enterica subsp. salamae
0.01 1 1 S2 2500152 Salmonella enterica subsp. salamae serovar 42:r:-
0.14 13 1 G 570 Klebsiella
0.09 8 7 S 573 Klebsiella pneumoniae
0.01 1 1 S1 72407 Klebsiella pneumoniae subsp. pneumoniae
0.01 1 1 S 244366 Klebsiella variicola
0.01 1 1 S 1134687 Klebsiella michiganensis
0.01 1 1 S 1463165 Klebsiella quasipneumoniae
0.01 1 1 S 2153354 Klebsiella sp. WCHKl090001
0.03 3 0 G 620 Shigella
0.01 1 1 S 621 Shigella boydii
0.01 1 1 S 622 Shigella dysenteriae
0.01 1 1 S 623 Shigella flexneri
0.02 2 0 F1 191675 unclassified Enterobacteriaceae
0.02 2 0 F2 84563 ant, tsetse, mealybug, aphid, etc. endosymbionts
0.01 1 0 F3 146507 aphid secondary symbionts
0.01 1 1 S 1199245 secondary endosymbiont of Ctenarytaina eucalypti
0.01 1 0 G 1906660 Candidatus Mikella
0.01 1 1 S 1778264 Candidatus Mikella endobia
0.01 1 0 G 547 Enterobacter
0.01 1 0 G1 354276 Enterobacter cloacae complex
0.01 1 1 S 158836 Enterobacter hormaechei
0.01 1 0 G 544 Citrobacter
0.01 1 0 G1 1344959 Citrobacter freundii complex
0.01 1 1 S 2066049 Citrobacter freundii complex sp. CFNIH2
0.01 1 0 G 1780190 Izhakiella
0.01 1 1 S 2579935 Izhakiella sp. KSNA2
7.40 672 0 F 1903411 Yersiniaceae
7.40 672 3 G 629 Yersinia
7.36 669 669 S 630 Yersinia enterocolitica
5.84 531 0 P1 68525 delta/epsilon subdivisions
5.84 531 0 C 29547 Epsilonproteobacteria
5.84 531 0 O 213849 Campylobacterales
5.84 531 0 F 72293 Helicobacteraceae
5.84 531 0 G 209 Helicobacter
5.84 531 530 S 210 Helicobacter pylori
0.01 1 1 S1 1163741 Helicobacter pylori Shi169
22.39 2035 0 D1 1783270 FCB group
22.39 2035 0 D2 68336 Bacteroidetes/Chlorobi group
22.39 2035 0 P 976 Bacteroidetes
22.39 2035 0 C 200643 Bacteroidia
22.39 2035 0 O 171549 Bacteroidales
22.39 2035 0 F 815 Bacteroidaceae
22.39 2035 0 G 816 Bacteroides
11.73 1066 1066 S 817 Bacteroides fragilis
10.65 968 271 S 821 Bacteroides vulgatus
7.67 697 697 S1 435590 Bacteroides vulgatus ATCC 8482
0.01 1 1 S 820 Bacteroides uniformis
9.84 894 0 P 32066 Fusobacteria
9.84 894 0 C 203490 Fusobacteriia
9.84 894 0 O 203491 Fusobacteriales
9.84 894 0 F 203492 Fusobacteriaceae
9.84 894 0 G 848 Fusobacterium
9.84 894 0 S 851 Fusobacterium nucleatum
9.84 894 379 S1 76856 Fusobacterium nucleatum subsp. nucleatum
5.67 515 515 S2 190304 Fusobacterium nucleatum subsp. nucleatum ATCC 25586
0.01 1 0 D1 1783257 PVC group
0.01 1 0 P 74201 Verrucomicrobia
0.01 1 0 C 203494 Verrucomicrobiae
0.01 1 0 O 48461 Verrucomicrobiales
0.01 1 0 F 1647988 Akkermansiaceae
0.01 1 0 G 239934 Akkermansia
0.01 1 1 S 239935 Akkermansia muciniphila
| Species | Strain | Species detected | Strain detected | Name of strain detected |
|---|---|---|---|---|
| Bacteriodes fragilis | ATCC 25285 | Yes | No | |
| Bacteroides vulgatus | ATCC 8482 | Yes | Yes | Bacteroides vulgatus ATCC 8482 |
| Clostridioides difficile | ATCC 9689 | Yes | Yes | Clostridioides difficile M68 |
| Enterobacter cloacae | ATCC 13047 | No | ||
| Escherichia coli | ATCC 700926 | Yes | Yes | Escherichia coli O7:K1 |
| Salmonella enterica subsp. enterica | ATCC 9150 | Yes | Yes | Salmonella enterica subsp. Enterica & Salmonella enterica subsp. Salamae |
| Bifidobacterium adolescentis | ATCC 15703 | Yes | Yes | Bifidobacterium adolescentis ATCC 15703 |
| Enterococcus faecalis | ATCC 700802 | Yes | No | |
| Lactobacillus plantarum | ATCC BAA-793 | Yes | No | |
| Helicobacter pylori | ATCC 700392 | Yes | Yes | Helicobacter pylori Shi169 |
| Yersinia enterocolitica | ATCC 27729 | Yes | No | |
| Fusobacterium nucleatum subsp. nucleatum | ATCC 25586 | Yes | Yes | Fusobacterium nucleatum subsp. nucleatum ATCC 25586 |
Of the 12 species in the sample, 11 could be detected to species level. Of these 11 detected species, seven could be detected to strain level. Of these seven detected strains, three are exact matches to the ATCC strain designation.
ATCC-oral
kraken2 --db Kraken2/standard_DB --threads 24 --out atcc_oral.kraken --report atcc_oral.kreport atcc_oral_noprimers_lengthfilter.fastq
Loading database information... done.
16173 sequences (23.61 Mbp) processed in 0.481s (2018.2 Kseq/m, 2945.82 Mbp/m).
16173 sequences classified (100.00%)
0 sequences unclassified (0.00%)
100.00 16173 0 R 1 root
100.00 16173 0 R1 131567 cellular organisms
100.00 16173 0 D 2 Bacteria
32.61 5274 0 D1 1783272 Terrabacteria group
28.76 4651 0 P 1239 Firmicutes
15.37 2485 0 C 91061 Bacilli
15.37 2485 0 O 186826 Lactobacillales
15.37 2485 0 F 1300 Streptococcaceae
15.37 2485 2450 G 1301 Streptococcus
0.10 16 16 S 2420308 Streptococcus sp. FDAARGOS_520
0.02 3 3 S 1346 Streptococcus iniae
0.02 3 3 S 1313 Streptococcus pneumoniae
0.01 2 0 G1 119603 Streptococcus dysgalactiae group
0.01 2 0 S 1334 Streptococcus dysgalactiae
0.01 2 0 S1 119602 Streptococcus dysgalactiae subsp. equisimilis
0.01 2 2 S2 486410 Streptococcus dysgalactiae subsp. equisimilis GGS_124
0.01 1 1 S 2420310 Streptococcus sp. FDAARGOS_522
0.01 1 1 S 1917441 Streptococcus ruminantium
0.01 1 1 S 150055 Streptococcus lutetiensis
0.01 1 1 S 28037 Streptococcus mitis
0.01 1 1 S 1319 Streptococcus sp. 'group B'
0.01 1 0 S 1311 Streptococcus agalactiae
0.01 1 1 S1 1309807 Streptococcus agalactiae ILRI005
0.01 1 1 S 1309 Streptococcus mutans
0.01 1 1 S 1308 Streptococcus thermophilus
0.01 1 1 S 1111760 Streptococcus troglodytae
0.01 1 1 S 1305 Streptococcus sanguinis
0.01 1 0 G1 671232 Streptococcus anginosus group
0.01 1 1 S 1338 Streptococcus intermedius
13.39 2166 0 C 909932 Negativicutes
13.39 2166 0 O 1843489 Veillonellales
13.39 2166 0 F 31977 Veillonellaceae
13.39 2166 0 G 29465 Veillonella
13.39 2166 2166 S 29466 Veillonella parvula
3.85 623 0 P 201174 Actinobacteria
3.85 623 0 C 1760 Actinobacteria
3.85 623 0 O 2037 Actinomycetales
3.85 623 0 F 2049 Actinomycetaceae
3.80 615 0 G 2529408 Schaalia
3.80 615 615 S 1660 Schaalia odontolytica
0.05 8 0 G 1654 Actinomyces
0.05 8 8 S 1852377 Actinomyces pacaensis
28.08 4542 0 P 32066 Fusobacteria
28.08 4542 0 C 203490 Fusobacteriia
28.08 4542 0 O 203491 Fusobacteriales
28.08 4542 0 F 203492 Fusobacteriaceae
28.08 4542 0 G 848 Fusobacterium
28.08 4542 0 S 851 Fusobacterium nucleatum
28.08 4542 2056 S1 76856 Fusobacterium nucleatum subsp. nucleatum
15.37 2485 2485 S2 190304 Fusobacterium nucleatum subsp. nucleatum ATCC 25586
0.01 1 1 S2 525283 Fusobacterium nucleatum subsp. nucleatum ATCC 23726
25.62 4144 0 P 1224 Proteobacteria
25.62 4144 0 C 1236 Gammaproteobacteria
25.62 4144 0 O 135625 Pasteurellales
25.62 4144 0 F 712 Pasteurellaceae
25.62 4144 0 G 724 Haemophilus
25.62 4144 4144 S 729 Haemophilus parainfluenzae
13.68 2213 0 D1 1783270 FCB group
13.68 2213 0 D2 68336 Bacteroidetes/Chlorobi group
13.68 2213 0 P 976 Bacteroidetes
13.68 2213 0 C 200643 Bacteroidia
13.68 2213 0 O 171549 Bacteroidales
13.68 2212 0 F 171552 Prevotellaceae
13.68 2212 0 G 838 Prevotella
13.68 2212 2212 S 28132 Prevotella melaninogenica
0.01 1 0 F 815 Bacteroidaceae
0.01 1 0 G 816 Bacteroides
0.01 1 1 S 817 Bacteroides fragilis
| Species | Strain | Species detected | Strain detected | Name of strain detected |
|---|---|---|---|---|
| Schaalia odontolytica | ATCC 17982 | Yes | No | |
| Prevotella melaninogenica | ATCC 25845 | Yes | No | |
| Fusobacterium nucleatum subsp. nucleatum | ATCC 25586 | Yes | Yes | Fusobacterium nucleatum subsp. nucleatum ATCC 25586 |
| Streptococcus mitis | ATCC 49456 | Yes | No | |
| Veillonella parvula | ATCC 17745 | Yes | No | |
| Haemophilus parainfluenzae | ATCC 33392 | Yes | No |
Of the six species in the sample, all six could be detected to species level. Of these six species detected, one could be detected to strain level, which was an exact match to the ATCC strain designation.
Madison Water Samples Analysis
To demonstrate the the application of LoopSeq 16S kit on real world environmental samples, four water samples were collected from Madison (Wisconsin, USA): 1) Home aquarium (Aquarium) 2) Feynman Pond outside the Promega office buildings (FeynmanPond) 3) Residential rain barrel collecting rain runoff (RainBarrel) 4) Riva Pond in a residential neighbourhood (RivaPond)
Samples were classified using an LCA classifer and diversity analysis was performed on the classification results.
The 16S + 18S kit was used but only the 16S contigs are examined.
Data Preparation
LoopSeq 16S Contig QC & Filtering
Madison-Aquarium
Load fastq file containing raw contigs as downloaded from the Loop Genomics cloud platform.
#R
library(dada2)
library(ShortRead)
F_prim <- "AGAGTTTGATCMTGGCTCAG" #forward primer
R_prim <- "TACCTTGTTACGACTT" #reverse primer
madison_aquarium <- file.path("madison_aquarium.fastq")
#Basic statistics of raw contigs
summary(nchar(getSequences(madison_aquarium)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
231 1393 1457 1355 1489 2921
#Remove forward and reverse primers. This step also ensures that the contigs that remain after filtering are the full length 16S gene.
removePrimers(madison_aquarium, "madison_aquarium_noprimers.fastq", primer.fwd=F_prim, primer.rev=rc(R_prim), compress=FALSE, max.mismatch=2, allow.indels=FALSE, verbose=TRUE)
6 sequences out of 18585 are being reverse-complemented.
Read in 18585, output 6967 (37.5%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed)
madison_aquarium_noprimers <- file.path("madison_aquarium_noprimers.fastq")
summary(nchar(getSequences(madison_aquarium_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1176 1442 1453 1443 1455 1544
#Still some contigs that are too short. Filter for length.
filterAndTrim(madison_aquarium_noprimers, "madison_aquarium_noprimers_lengthfilter.fastq", minLen=1400, maxLen=1600, verbose=TRUE, compress=FALSE)
Read in 6967, output 6428 (92.3%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed, length filtered)
madison_aquarium_noprimers_lengthfilter <- file.path("madison_aquarium_noprimers_lengthfilter.fastq")
summary(nchar(getSequences(madison_aquarium_noprimers_lengthfilter)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1402 1451 1453 1447 1455 1544
FeynmanPond
Load fastq file containing raw contigs as downloaded from the Loop Genomics cloud platform.
#R
library(dada2)
library(ShortRead)
F_prim <- "AGAGTTTGATCMTGGCTCAG" #forward primer
R_prim <- "TACCTTGTTACGACTT" #reverse primer
madison_feynman_pond <- file.path("madison_feynman_pond.fastq")
#Basic statistics of raw contigs
summary(nchar(getSequences(madison_feynman_pond)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
222 1376 1451 1352 1487 2714
#Remove forward and reverse primers. This step also ensures that the contigs that remain after filtering are the full length 16S gene.
removePrimers(madison_feynman_pond, "madison_feynman_pond_noprimers.fastq", primer.fwd=F_prim, primer.rev=rc(R_prim), compress=FALSE, max.mismatch=2, allow.indels=FALSE, verbose=TRUE)
5 sequences out of 20189 are being reverse-complemented.
Read in 20189, output 8251 (40.9%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed)
madison_feynman_pond_noprimers <- file.path("madison_feynman_pond_noprimers.fastq")
summary(nchar(getSequences(madison_feynman_pond_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1376 1434 1446 1442 1457 1522
RainBarrel
Load fastq file containing raw contigs as downloaded from the Loop Genomics cloud platform.
#R
library(dada2)
library(ShortRead)
F_prim <- "AGAGTTTGATCMTGGCTCAG" #forward primer
R_prim <- "TACCTTGTTACGACTT" #reverse primer
madison_rain_barrel <- file.path("madison_rain_barrel.fastq")
#Basic statistics of raw contigs
summary(nchar(getSequences(madison_rain_barrel)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
204 1337 1443 1349 1482 2671
#Remove forward and reverse primers. This step also ensures that the contigs that remain after filtering are the full length 16S gene.
removePrimers(madison_rain_barrel, "madison_rain_barrel_noprimers.fastq", primer.fwd=F_prim, primer.rev=rc(R_prim), compress=FALSE, max.mismatch=2, allow.indels=FALSE, verbose=TRUE)
2 sequences out of 15140 are being reverse-complemented.
Read in 15140, output 4673 (30.9%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed)
madison_rain_barrel_noprimers <- file.path("madison_rain_barrel_noprimers.fastq")
summary(nchar(getSequences(madison_rain_barrel_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1375 1436 1446 1443 1456 1496
RivaPond
Load fastq file containing raw contigs as downloaded from the Loop Genomics cloud platform.
#R
library(dada2)
library(ShortRead)
F_prim <- "AGAGTTTGATCMTGGCTCAG" #forward primer
R_prim <- "TACCTTGTTACGACTT" #reverse primer
madison_riva_pond <- file.path("madison_riva_pond.fastq")
#Basic statistics of raw contigs
summary(nchar(getSequences(madison_riva_pond)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
242 1090 1412 1253 1448 2036
#Remove forward and reverse primers. This step also ensures that the contigs that remain after filtering are the full length 16S gene.
removePrimers(madison_riva_pond, "madison_riva_pond_noprimers.fastq", primer.fwd=F_prim, primer.rev=rc(R_prim), compress=FALSE, max.mismatch=2, allow.indels=FALSE, verbose=TRUE)
7 sequences out of 14352 are being reverse-complemented.
Read in 14352, output 3808 (26.5%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed)
madison_riva_pond_noprimers <- file.path("madison_riva_pond_noprimers.fastq")
summary(nchar(getSequences(madison_riva_pond_noprimers)))
Min. 1st Qu. Median Mean 3rd Qu. Max.
1350 1410 1419 1428 1449 2000
#Still some contigs are too long. Filter for length.
filterAndTrim(madison_riva_pond_noprimers, "madison_riva_pond_noprimers_lengthfilter.fastq", minLen=1400, maxLen=1600, verbose=TRUE, compress=FALSE)
Overwriting file:C:\Users\Nico\Documents\workdir\madison_riva_pond_noprimers_lengthfilter.fastq
Read in 3808, output 3564 (93.6%) filtered sequences.
#Basic statistics of filtered contigs (full length 16S, primers trimmed, length filtered)
madison_riva_pond_noprimers_lengthfilter <- file.path("madison_riva_pond_noprimers_lengthfilter.fastq")
summary(nchar(getSequences(madison_riva_pond_noprimers_lengthfilter)))
Classification with LCA Classifier
Taxonomic classification with Kraken 2 outputs reports which show a hierarchical LCA scheme. While most full length 16S contigs can be classified to species level, some contigs can only be classifed to higher taxonomic levels. To obtain species abundances, the companion tool Bracken is run to redistribute all contigs to species level. This redistribution is limited to lineages with at least one species identified. While this method may not fully represent the species diversity in the sample, it does allow a comparison of relative abundances of species that can be classified.
Aquarium
kraken2 --db Kraken2/standard_DB --threads 24 --out madison_aquarium.kraken --report madison_aquarium.kreport madison_aquarium_noprimers_lengthfilter.fastq
Loading database information... done.
6428 sequences (9.30 Mbp) processed in 0.440s (876.8 Kseq/m, 1268.39 Mbp/m).
6428 sequences classified (100.00%)
0 sequences unclassified (0.00%)
bracken -d Kraken2/standard_DB -i madison_aquarium.kreport -o madison_aquarium.bracken -r 1500 -l S -t 1
>> Checking for Valid Options...
>> Running Bracken
>> python src/est_abundance.py -i madison_aquarium.kreport -o madison_aquarium.bracken -k Kraken2/standard_DB/database1500mers.kmer_distrib -l S -t 1
PROGRAM START TIME: 12-31-2019 05:36:30
BRACKEN SUMMARY (Kraken report: madison_aquarium.kreport)
>>> Threshold: 1
>>> Number of species in sample: 118
>> Number of species with reads > threshold: 118
>> Number of species with reads < threshold: 0
>>> Total reads in sample: 6428
>> Total reads kept at species level (reads > threshold): 6129
>> Total reads discarded (species reads < threshold): 0
>> Reads distributed: 68
>> Reads not distributed (eg. no species above threshold): 231
>> Unclassified reads: 0
BRACKEN OUTPUT PRODUCED: madison_aquarium.bracken
PROGRAM END TIME: 12-31-2019 05:36:31
Bracken complete.
FeynmanPond
kraken2 --db Kraken2/standard_DB --threads 24 --out madison_feynman_pond.kraken --report madison_feynman_pond.kreport madison_feynman_pond_noprimers.fastq
Loading database information... done.
8251 sequences (11.89 Mbp) processed in 0.516s (959.3 Kseq/m, 1382.90 Mbp/m).
8251 sequences classified (100.00%)
0 sequences unclassified (0.00%)
bracken -d Kraken2/standard_DB -i madison_feynman_pond.kreport -o madison_feynman_pond.bracken -r 1500 -l S -t 1
>> Checking for Valid Options...
>> Running Bracken
>> python src/est_abundance.py -i madison_feynman_pond.kreport -o madison_feynman_pond.bracken -k Kraken2/standard_DB/database1500mers.kmer_distrib -l S -t 1
PROGRAM START TIME: 12-31-2019 05:45:10
BRACKEN SUMMARY (Kraken report: madison_feynman_pond.kreport)
>>> Threshold: 1
>>> Number of species in sample: 332
>> Number of species with reads > threshold: 332
>> Number of species with reads < threshold: 0
>>> Total reads in sample: 8251
>> Total reads kept at species level (reads > threshold): 5870
>> Total reads discarded (species reads < threshold): 0
>> Reads distributed: 496
>> Reads not distributed (eg. no species above threshold): 1885
>> Unclassified reads: 0
BRACKEN OUTPUT PRODUCED: madison_feynman_pond.bracken
PROGRAM END TIME: 12-31-2019 05:45:10
Bracken complete.
RainBarrel
kraken2 --db Kraken2/standard_DB --threads 24 --out madison_rain_barrel.kraken --report madison_rain_barrel.kreport madison_rain_barrel_noprimers.fastq
Loading database information... done.
4673 sequences (6.74 Mbp) processed in 0.274s (1022.6 Kseq/m, 1475.63 Mbp/m).
4673 sequences classified (100.00%)
0 sequences unclassified (0.00%)
bracken -d Kraken2/standard_DB -i madison_rain_barrel.kreport -o madison_rain_barrel.bracken -r 1500 -l S -t 1
>> Checking for Valid Options...
>> Running Bracken
>> python src/est_abundance.py -i madison_rain_barrel.kreport -o madison_rain_barrel.bracken -k Kraken2/standard_DB/database1500mers.kmer_distrib -l S -t 1
PROGRAM START TIME: 12-31-2019 06:28:32
BRACKEN SUMMARY (Kraken report: madison_rain_barrel.kreport)
>>> Threshold: 1
>>> Number of species in sample: 165
>> Number of species with reads > threshold: 165
>> Number of species with reads < threshold: 0
>>> Total reads in sample: 4673
>> Total reads kept at species level (reads > threshold): 2336
>> Total reads discarded (species reads < threshold): 0
>> Reads distributed: 1489
>> Reads not distributed (eg. no species above threshold): 848
>> Unclassified reads: 0
BRACKEN OUTPUT PRODUCED: madison_rain_barrel.bracken
PROGRAM END TIME: 12-31-2019 06:28:32
Bracken complete.
RivaPond
kraken2 --db Kraken2/standard_DB --threads 24 --out madison_riva_pond.kraken --report madison_riva_pond.kreport madison_riva_pond_noprimers_lengthfilter.fastq
Loading database information... done.
3564 sequences (5.10 Mbp) processed in 0.339s (631.5 Kseq/m, 903.08 Mbp/m).
3564 sequences classified (100.00%)
0 sequences unclassified (0.00%)
bracken -d Kraken2/standard_DB -i madison_riva_pond.kreport -o madison_riva_pond.bracken -r 1500 -l S -t 1
>> Checking for Valid Options...
>> Running Bracken
>> python src/est_abundance.py -i madison_riva_pond.kreport -o madison_riva_pond.bracken -k Kraken2/standard_DB/database1500mers.kmer_distrib -l S -t 1
PROGRAM START TIME: 12-31-2019 06:16:39
BRACKEN SUMMARY (Kraken report: madison_riva_pond.kreport)
>>> Threshold: 1
>>> Number of species in sample: 166
>> Number of species with reads > threshold: 166
>> Number of species with reads < threshold: 0
>>> Total reads in sample: 3564
>> Total reads kept at species level (reads > threshold): 2507
>> Total reads discarded (species reads < threshold): 0
>> Reads distributed: 87
>> Reads not distributed (eg. no species above threshold): 970
>> Unclassified reads: 0
BRACKEN OUTPUT PRODUCED: madison_riva_pond.bracken
PROGRAM END TIME: 12-31-2019 06:16:39
Bracken complete.
Diversity Analysis
The number of species identified and top 3 species by abundance.
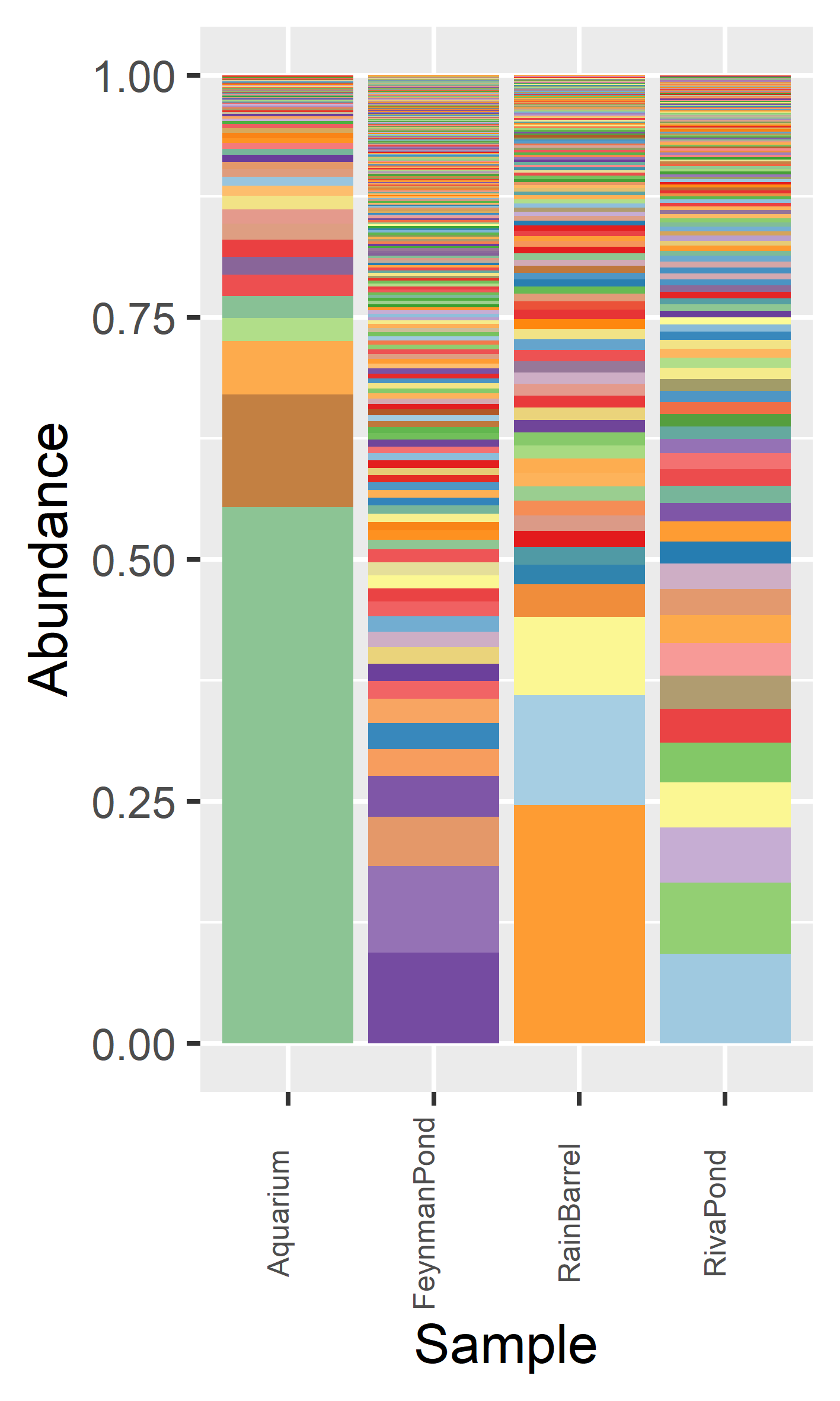
| Sample | Species count | First | Second | Third |
|---|---|---|---|---|
| Aquarium | 118 | Limnohabitans sp. 63ED37-2 (0.55) | Polynucleobacter necessarius (0.12) | Novosphingobium ginsenosidimutans (0.05) |
| FeynmanPond | 332 | Candidatus Planktophila limnetica (0.09) | Arachidicoccus ginsenosidivorans (0.09) | Limnohabitans sp. 103DPR2 (0.05) |
| RainBarrel | 165 | Steroidobacter denitrificans (0.25) | Pseudomonas sp. SWI6 (0.11) | Planctomycetes bacterium Pan189 (0.08) |
| RivaPond | 166 | Scytonema sp. HK-05 (0.09) | Niabella soli (0.07) | Planctomycetes bacterium Pan97 (0.06) |
Combine all bracken reports into a contingency table, convert to biom file and run plot rank abundance.
biom convert -i madison_water_combined.txt -o madison_water_combined.biom --to-hdf5 --table-type="OTU table" --process-obs-metadata taxonomy
plot_rank_abundance_graph.py -i madison_water_combined.biom -s '*' -x -f png -v -o madison_water_combined_rank_abundance.png
![]()
The Aquarium sample is dominated by fewer high abundance species while the FeynmanPond sample is most diverse and with a more even species distribution.
#R
library(dplyr)
library(ggplot2)
library(RColorBrewer)
#Melt and add sample info to madison_water_combined.txt
data <- read.csv("madison_water_combined.csv", header = TRUE, stringsAsFactors = FALSE)
data <- data %>% arrange(Sample, Abundance)
x <- ggplot(data, aes(x=Sample, y=Abundance, fill=factor(Species),group=Abundance)) +
geom_bar(stat="identity",position="stack") +
theme(legend.position="none",
axis.text.x = element_text(size=10,angle=90,vjust=0))
cols <- colorRampPalette(brewer.pal(12, "Paired"))
myPal <- cols(length(unique(data$Species)))
x + scale_fill_manual(values = sample(myPal))
newpath <- paste0("madison_water_combined_barchart.png")
ggsave(filename=newpath,width=3,height=8,units="cm",dpi=600)